ABSTRACT:
Promoter reporter system monitors the gene expression in many organisms. The reporter gene helps to identify molecular events and gene transcription, which promotes the many research of the molecular- basis studies of different diseases and new technology on drug development. To test the activity of this system, we designed a recombinant plasmid contains an inserted foreign T7 promoter region upstream to the Green Fluorescent Protein reporter gene in the pGFP-S12, and then expect the GFP reporter gene expression will be restored by this event. The T7 promoter found on the pGEM-T was cloned via PCR and then inserted to pGFP-S12 via ligation. The ligation product was transformed into DH5α strain E. coli. After selection, the recombinant plasmid of interest was screened on gel electrophoresis after restriction endonucleases. Our results from the plasmid screen indicated that the structure of the recombinant plasmid was not the same as we expected. If the recombinant plasmids are successfully produced, then they can be carry on to further studies to test the activity of T7 promoter reporter system.
INTRODUCTION:
The reporter gene has the ability to “report”, due to its highly controllable, sensitive, reliable and convenient quality in large-scale experiment, so they are widely used in molecular biology to monitor gene expression in cellular activities in many biological organisms (Naylor, 1999). Moreover, because of its unique phenotypic expression of reporter genes, the reporter gene can visually help to provide evidence to support or disprove any scientific hypothesis (Brogan et al., 2012). Nowadays, one of the most popular reporter gene is the green fluorescent protein (gfp) found in Aequorea aequorea (Reed et al., 2001). This reporter gene has been commonly in many experiments (Heifetz, 2000), one reason is that this gene has a very special phenotype of fluoresce that can be easily distinguished when exposed under UV light. Scientist can often determine whether their result is supportive or against their theory based on the observation of fluorescent event of this reporter gene. another reason is the GFP reporter gene can be used in vivo experiment, which allows scientist to visualize the cell division and cell growth within cells (Hoffman, 2015). Study cell division and cell growth process are very important in molecular biology, one of the famous applications is the study of cell division in cancer research. Cancer often result from uncontrollable division of cells (Pike et.al, 1993). By using the reporter gene, the cell division can be easily tracked, therefore, cancer studies can be conducted.
The expression of GFP reporter gene can be controlled by a promoter reporter system. A promoter upstream of the GFP reporter gene can restore the transcription of this gene, thereby control the protein synthesis and limit its fluorescent ability. Whether the fluorescence present or not can reflect the promoter’s function and ability in controlling gene expression. And this generally assess the promoter’s activity. (Soboleski, Oaks and Halford, 2005). Kunert et.al. (2000) used GFP reporter gene in Cyanobacterium Synechocystis sp. strain PCC 6803 to assess the activity of the promoter. The interaction between the prompters and the reporter genes can be useful in detecting the changes in expression of reporter gene that induced by different promoter activity levels. For example, Thomas et.al. (1989) found that firefly luciferase (LUC) reporter gene is extremely helpful in study interleukin-2 (IL2) promoter, they tested the difference between light emission level of the transfected cell to monitor the promoter’s activity. This system is also widely used in laboratory to test the promoter or reporter gene function in plasmid. In addition, Goh et al. (2002) successfully cloned promoter-lux reporter into a plasmid of Salmonella typhimurium and tested the gene expression response. Their findings also showed that promoter reporter systems can be used to investigate promoter activity.
In Jan. 2012, students from the University of British Columbia constructed a GFP reporter plasmid by removing the lacZ promoter form the pGFPuv plasmid. The newly-generated plasmid was named pGFP-S12 and its structure and sequence were confirmed in the following summer by other students. The purpose of this experiment is to identify whether pGFP-S12 reporter plasmid can successfully “report” the inserted promoter’s function and ability, and we expect to find similar result as described in Goh et al.’s (2002) study that the inserted promoter can change the reporter gene expression. To be specific, pGFP-S12 reporter plasmid will be inserted with a T7 promoter, and based on the expression of the GFP gene, we can analyze the role of this promoters in restoring gene expression. However, the promoter’s function and ability cannot be determined if the insertion of promoter fails. And this may lead to a further question of whether pGFP-S12 is a qualified reporter plasmid for testing the promoter reporter system.
To test the function of the inserted promoter and the reporter plasmid, we cloned the T7 promoter region from pGEM-T, and then ligated this region into the pGFP-S12 plasmid. We hypothesize that with the presence of T7 RNA polymerase, GFP reporter gene can successfully “report” the function of the T7 promoter region by presenting fluoresces, thus proves the pGFP-S12 plasmid can be used in studying promoter reporter system. To test our hypothesis, we cloned and amplified the T7 promoter region from pGEM-T using LCT7 primer via PCR and ligated it with pGFP-S12. The newly-generated recombinant plasmid was then transformed into DH5α Escherichia coli cells, and then only successful ligation products were selected to test whether the insertion of promoter happened or not. However, due the fact of DH5α E. coli cells used in this experiment do not have T7 RNA polymerase, therefore the selection of ligation product will not fluorescent. This may affect the “report “ ability of reporter gene, thus further confirmation of this experiment is needed.
MATERIALS AND METHODS
All materials and equipment were provided by the University of British Columbia Biology 341 laboratory.
Plasmid identification.
Restriction digests were performed by following the instruction of Chen (2019). The aim of this step is to confirm the identity of unknown plasmid X. This potential identity of this plasmid could be either pGFP-S12 or pGEM-T. 1μg of plasmid X was digested using restriction enzymes BamHI, PvuII-HF and NdeI with CutSmart Buffer in a total volume of 15μL and were incubated at 37°C for 90 minutes. DNA fragments sizes were examined and visualized using gel electrophoresis on a 0.8% agarose gel with the Invitrogen 1Kb Plus DNA marker used as a ladder.
Polymerase chain reaction (PCR).
PCR was performed according to Chen (2019), the purpose of this step is to amplify and add sticky ends to the T7 promoter region, so it can be cloned form pGEM-T and prepared for ligation in later step. The amplification involved usage of two primer sets: LCT7 designed by Liane Chen (the forward primer has the sequence of GATGAC AGATCT GCG CGT AAC CAC CAC ACC, and the reverse primer has the sequence of TAGATC AGATCT CGG GAG CAT GCG ACG TCG ), and ME/RB designed by Masha Eslami and Ritika Bhinder (the forward primer has the sequence of AGT TCG GGA TCC CTC TTC GCT ATT ACG CCA G, and the reverse primer has the sequence of AGT TCG GGA TCC CAC AGG AAA CAG CTA TGA CC). Primer set LCT7 and ME/RB were added with pGEM-T separately to perform PCR reaction. Then, PCR products were visualized by running them on a Lonza FlashGelTM using gel electrophoresis, as indicated by Chen (2019), and a standard FlashGelTM DNA marker was used as ladder. Each PCR reaction has compared to no DNA template control to test the if there is contamination.
Later, the purification of the PCR products was performed according to QIAQuick PCR Purification Kit handbook (2019) using column purification method. Then, the purity and concentration of the isolated PCR products was examined on NanoDrop Microvolume Spectrophotometer.
Ligations and transformations.
To prepare for the ligation, 4 μL of 0.5 μg/μL pGFP-S12 plasmid was digested using BglII to activate its sticky ends and incubated at 37°C for 2 hours and 30 minutes (Chen, 2019). Then, 30μL of LCT7 PCR product was digested using BglII and incubated at 37°C for only 46 minutes, which is slightly less than the required incubation time as Chen described (2019). The isolation of DNA and inactivation of excess enzymes were performed as described in the QiaQuick PCR Purification Kit Handbook (2019).
According to the ligation methods from Chen (2019), a 1:3 vector to insert ratio ligation and a 1:10 ratio vector to insert ratio ligation were performed using pGFP-S12 reporter plasmid and T7 promoter region (from the clean PCR product).
Transformation was also performed as Chen outlined (2019). 1:3 ligation product and 1:10 ligation product were transformed with DH5α E. coli cells. And then transferred and evenly spread on LB-agar-ampicillin plates and incubated in suitable environment at 37°C for 12 hours. Also, another two control groups were set up as Chen (2019) required: pGFP-S12 plasmid transformed with DH5α E. coli cells and plated on ampicillin presenting plate, pGFPuv plasmid transformed with DH5α E. coli cells and plated on ampicillin presenting plate. In addition, two control group were performed by Ryan Ghorayeb and Elizabeth Samuels: untransformed DH5α E. coli cells plated on no ampicillin plate, and untransformed E. coli cells plated on ampicillin presenting plate. The result of bacterial growth and fluorescence were checked under a UV transilluminator.
Small scale plasmid prep and quantification.
According to Chen (2019), only the recombinant plasmid from a successful ligation can be used to test the insertion of T7 promoter. The successfully transformed cultures (no fluorescent and no satellite colonies growing on its side) were picked from each transformation plates (2 cultures form 1:3 ligation plates and 2 cultures from 1:10 ligation plates). The bacteria cultures were inoculated with LB-Ampicillin (50μg/mL) and incubated at 37°C for 12 hours. Then the plasmid DNA was isolated according to the QIAprep Spin Miniprep Kit Handbook (2019) using the column purification method. Then, the purity and concentration of the isolated PCR products were examined on NanoDrop Microvolume Spectrophotometer.
Plasmid Screening:
To verify the the insertion of T7 region to pGFP-S12, a restriction endonuclease was performed according to Chen (2019). Restriction digest using enzymes BglII to analyze the fragment size of recombinant plasmid (pGFP-S12 plus T7 region), the digestion was incubated for two hours at 37°C. Finally, gel electrophoresis was used to visually present the result, the digested samples were run on 1.0% agarose gel, and a 1Kb Plus DNA ladder was used as an indicator to show fragment size.
RESULTS
Restriction Analysis of Identification of Plasmid X
Restriction digestion of pGFP-S12 and pGEM-T with enzymes: BamHI, NdeI and PvuII, can produce distinct band patterns on gel. Thus, those enzymes were selected to verify the identity of plasmid X, and gel electrophoresis was used to display the result. The maps of the respective plasmids (Fig 1 b&c) visually explained the enzyme digest sites and the resultant fragment sizes of the two plasmids (Fig 1 b& c). Three digests were performed, the predicted fragment size and restriction sites for both pGFP-S12 and pGEM-T are showed in figure 1a. Restriction digest result of plasmid X was shown on the gel as in Fig 1d, fragments size were determined by comparison with 1kb Invitrogen DNA ladder in Lane 1. The uncut plasmid in control group displayed two obvious supercoiled bands corresponding to approximately 2.3kb and 1.6kb (Fig 1d). Also, the digestion took place with PvuII produced similar results of fragment size in Lane 5 with one supercoiled band range from 2.3kb to1.6kb, which indicates that this plasmid has not been cut by PvuII. In addition, digestion with BamHI resulted in fragments of approximately 2.4kb and 0.5kb (Fig 1d), and digestion with NdeI produced fragments of approximately 2.0kb and 0.8kb (Fig 1d). Above results indicated that the identity of plasmid X is pGFP-S12, since all bands and their sizes are closely related to the prediction in Fig 1a of pGFP-S12 (with predicted fragment sizes of 2.5kb and 0.5kb cut by BamHI and predicted fragment sizes of 2.3kb and 0.7kb cut by NdeI). In contrast, all the evidence above showed that it is impossible that plasmid X would be pGEM-T, since pGEM-T is predicted to have a single 3.0kb fragment when NdeI added and two fragments at 0.4kb and 2.5kb when PvuII added, and remain uncut when BamHI added (Fig 1a). The gel electrophoresis result showed that the band pattern was completely different from the prediction of band pattern of pGEM-T. Therefore, plasmid X is identified to be pGFP-S12.
Polymerase Chain Reaction (PCR):
After confirming the identity of plasmid X, sticky ends (restriction sites) have been added to two ends of the T7 promoter region to enhance the ability of holding the two pieces of DNA together in later ligation step when DNA ligase added. In order to add restriction enzymes sites to the two ends of the T7 promoter region, the polymerase chain reaction (PCR) using two sets of primers, LCT7 and ME/RB, has been performed. Then the PCR product samples have been visualized on Lonza FlashGel.
After undergoing amplification with the LCT7 primer, the PCR product with LCT7 primer showed a single band of approximately 300 bp in lane 2(Fig. 2), which is consistent with the prediction of the T7 promoter region has the fragment size of 268 bp. In addition, the control sample without DNA template and added LCT7 primer produced a smear band ranging about 300 bp. to 500 bp. In lane 1, this faint smear band indicated that there is DNA contamination during the process. The smear suggests that the contamination may be due to the undiluted sample of the PCR product with LCT7 primer accidentally fell into the well.
Meanwhile, amplification of pGEM-T using the ME/RB primer produced a single band of approximately 300 bp. in lane 5 (Fig. 2), which is consistent with the predicted digested product size of 314 bp. The control sample without DNA template using ME/RB primer produced no band on the gel (Fig. 2), which suggests that there is no DNA contamination during the process and the primer worked well.
The results on the gel showed that the PCR with LCT7 primer and ME/RB primer successfully amplified the T7 promoter region of the pGEM-T plasmid and also successfully added restriction sites to this region, though contamination could happen during this process. This result confirmed that the PCR product can be carried to the ligation and transformation step.
Transformation:
The purpose of the transformation step here was to select the recombinant plasmid that have the possibility of having a T7 promoter inserted, the colonies that contain the desired recombinant plasmid were expected to be no fluoresces and no satellite colonies growing around them. After the ligation, the ligation products were transformed into DH5α E. coli cells and plated on LB-agar-ampicillin plates, pGFPuv transformed into DH5α E. coli cells and pGFP-S12 that transformed into DH5α E. coli cells and plated on LB-agar-ampicillin plate and LB-agar plate were also used to show difference. The growth of cultures was examined under UV light. As observed, in one of the control sample, pGFPuv transfected cells (Fig. 3C) produced about 9 colonies and exhibited fluorescent as expected. E. coli cells transformed with pGFP-S12 showed large amount of colony growth in the LB-agar-ampicillin plate (Fig. 3D), which suggested that this plasmid contains an ampicillin resistance gene. Untransfected E. coli exhibited no growth on LB-agar-ampicillin (Fig. 3E) and untransfected E. coli on LB-agar without ampicillin also exhibited no growth, (Fig. 3F). The results here suggested that those E. coli cells are viable.
Since E. coli does not express the T7 polymerase, so it cannot initiate the T7 promoter and drive GFP expression within cells. In contrast, the observed colonies form Fig.3A and 3B, did not fluorescent as expected. There were multiple colonies growing on the plates, which indicated the ligation product of pGFP-S12 of 1:3 vector to insert ratios and 1:10 vector to insert ratios should contain a functional ampicillin resistance gene. The 1:3 vector to insert ratio ligation product produced approximately 21 colonies (Fig. 3A), and the 1:10 vector to insert ratio ligation product produced approximately 22 colonies (Fig. 3B).
In conclusion, these results above suggest that the ligation and transformation step is successfully done, and colonies can be produced in one of our ligation plates.
Plasmid Screening
After the potential recombinant plasmid DNA was selected from the E. coli transformation cells and the purified, we then screened it to confirm the whether there is a insertion of the T7 promoter region. Restriction digest using BglII and NEBuffer 3.1 was performed to determine whether the plasmid form transformed bacteria culture has pGFP-S12 or recombinant plasmid.
Based on figure 4a, the uncut pGFP-S12 (on lane 1) produced two bands at size around 3 kb and 12 kb. The uncut plasmid produced a dominant bright 3kb band which was identical to the bright band of size approximately 3kb that produced by pGFP-S12 digested with BglII in lane 2. This result indicates that this plasmid was supercoiled. And this result compatible with prediction of an uncut plasmid, and the result of lane 2 was also consistent with our prediction. However, digestion with our 1:3 ligated recombinant plasmid and 1:10 ligated recombinant plasmid did not show different banding patterns compared to lane 2. As observed form lane 3 to 6, all of the restriction digest results only showed a single band at approximately 3 kb and no other bands presented. This result contradicted with our prediction of BgIII will cut pGFP-S12 twice and produced two fragments that have a size of 3140 bp and 266 bp. This result suggested that all four colonies that we tested may not contain the inserted T7 region.
Having determined that our recombinant plasmid did not have inserted region, we then looked at recombinant plasmids from Jade and Emma’s restriction digest result. They used enzyme NdeI to test if their recombinant plasmid has a correct structure (Fig 4b), since they used a ME/RB primer set to do their PCR. Their uncut plasmid pGFP-S12 produced a dominant supercoiled band of approximately 2 kb, which is consistent with the prediction. The pGFP-S12 digested with NdeI only displayed a smear band on the gel. This result did not correspond to the prediction of two fragments of 2376 bp and 764 bp. This may result from an incomplete digestion. The recombinant plasmid containing a single T7 region inserted was predicted to have three fragments of 426 bp, 764 bp and 1624 bp when digested by NdeI. In lane 3,4 and 6, the digested results displayed 2 bands at approximately 800 bp and 1.6 kb. Those two bands is consistent with the prediction of fragment size of 764 bp and 1624 bp. However, the absence of the 426 bp band showed that there might be no T7 region inserted to the recombinant plasmid tested in these lanes. In lane 5, only a single smear band of approximately 2 kb to 5 kb presented on the gel, which indicates that there was an unsuccessful digestion or dilution.
All the above data lead to the conclusion that there might be no T7 region inserted into the pGFP-S12 reporter plasmid, thus we cannot perform further study on these failed recombinant DNA samples.
DISCUSSION
Whether the expression of the reporter gene is successful or not is determined by how this report gene interact with its promoters. This interaction is controlled by where to place the promoter and whether there is an appropriate polymerase to initiate the promoter’s function. With presence of corresponding polymerase, the insertion of promoter has to be located upstream of a promoter-less reporter gene, so that the promoter can restore the gene expression. This project has been designed to test the hypothesis of the GFP reporter gene in pGFP-S12 reporter plasmid can successfully express the reporter when a T7 promoter was inserted to the upstream of it. Our experiments aimed to prove this hypothesis.
Based on the result of PCR product verification on Lonza FlashGel, both primer sets successfully bind and amplified the desired region on pGEM-T as expected. However, the PCR reaction with the LCT7 primer set showed contamination in the no template control lane. As we can observe, the band pattern of the contamination was similar to the band pattern of the PCR product produced with LCT7 primer set, therefore, we can conclude that the DNA contamination is due to the unintentionally transferred PCR product in the control lane. And this contamination was not due the PCR procedure. As Scherczinger et al. suggested (1999), the false operation in handling the samples and transferring the samples can cause contamination. As observed, only a very faint band is shown on the control lane, which indicates only a small amount of DNA entered the sample. The PCR is extremely sensitive, a small amount of unwanted DNA could be amplified so many times and produced enough amount to be shown on the gel. If the contamination enters the sample before PCR reaction, the plasmid will be amplified exponentially during the reaction, and as a result, a brighter band should be presented on the gel. In addition, this contamination may due to the contaminated reagent, since we cannot make sure the environment is clean. Foreign DNA samples such as dead skin cells or saliva may enter our samples. Also, this contamination can be result from the formation of primer dimer. Although our PCR reactions using the LCT7 primer set were slightly contaminated, this contamination occurred after the PCR reaction, thus, we can conclude that our PCR products can be carried to next step. In addition, the result of PCR reaction with the ME/RB primer showed no contamination happened during the process. This successful reaction also allowed us to continue with our experiment.
The transformation result were as expected, the colonies with recombinant plasmid that grew on the LB-agar-ampicillin plates are non-fluorescent under UV luminescence. The recombinant plasmid was predicted to contain a functional T7 promoter region and a GFP reporter gene, but due to the DH5α strain of E. coli are lack of T7 RNA polymerase, the reporter gene still cannot “report” the promoter’s function (Tabor, 1990). In contrast, the GFP reporter gene in pGFPuv plasmid do not require T7 RNA polymerase, since its expression is controlled by the lacZ promoter and polymerase system (Karunakaran et.al., 2005), therefore it can be expressed and fluoresces can be observed when transformed with DH5α E. coli strain. Bacteria cells contain an ampicillin resistance gene can successfully grow on LB-agar-ampicillin plates, otherwise ampicillin will bind to the penicillin-binding-proteins in E. coli and inhibit cell wall synthesis and lead to cell death (Waxman and Strominger, 1983).
Also, control plates from Ryan Ghorayeb and Elizabeth Samuels show results consistent with predictions. This result may indicate that a single T7 promoter has been successfully insert to upstream of the GFP gene in pGFP-S12 reporter plasmid. However, this result only provide support to our hypothesis, but it cannot prove whether pGFP-S12 reporter plasmid is a successful plasmid. So, further investigation is needed, we should confirm GFP reporter gene activity by isolating the recombinant plasmid from our component cells and transformed into another strain of E. coil. or another bacteria that has T7 RNA polymerase, so that we can observe the promoter reporter system’s function. We proposed a potential candidate, strain BL21 E. coli, which contains the T7 RNA polymerase that can provide support to the T7 promoter reporter system’s function (Zawadzki and Gross, 1991). When the recombinant plasmid transformed with the presence of T7 RNA polymerase in BL21 E. coli, we expect to observe fluorescent under UV illumination, indicating that the insertion of promoter can initiate the reporter gene expression.
Plasmid screening of our ligation product produced an inconclusive result which indicates that our evidence is insufficient to prove that our ligations successfully linked the T7 promoter region to pGFP-S12 plasmid. The similarity between the recombinant plasmid and the pGFP-S12 indicated that there is no T7 promoter inserted to the plasmid, thereby proved there may be some error in our ligation. Thus, we cannot state the result is supportive to our hypothesis. All of our restriction digest results on gel displayed a single thick bended band, which indicates the DNA samples are supercoiled. And our bands were very bright, which indicates there were excess amount of DNA sample in the well. The excess amount of DNA may because we used 2μg of our ligation product in our sample. Also, the topmost faint bands presented in most lanes may result from the formation of nicked plasmid. This may due to the unexpected cutting from the introduced enzyme. All the above reason suggest that we may had used an improper functioning restriction enzyme in our analysis. On the other hand, recombinant plasmids’ structure can be identified without use of restriction enzymes, the method is called “blue/white screening” (Chaffin and Rubens, 1998). The disrupted plasmid (plasmid with an insert region) will be white, and the no insert DNA will be blue. This method will provide a quick analysis of the ligation success. If new ligation will be performed, then we can get the desired recombinant plasmid and carry it to further study to check if the insert is in the right direction.
Similar results can also be found in comparable studies in investigating T7 promoter’s activity. According to Tabor and Richardson’s bacteriophage experiment (1985), T7 RNA polymerase gene was inserted into plasmid pBR322, the result showed that specificity of T7 RNA polymerase for its own promoters (in this case lambda PL promoter) and the inhibitory ability with rifampicin allow the exclusive genes expression under the control of a T7 RNA polymerase and promoter system. In addition, T7 RNA polymerase activity can be tested in not only the bacteria cells but also in mammalian cells (Ghaderi et al., 2014). They introduced an Internal Ribosome Entry Site (IRES) sequences in mammalian cell to help the foreign gene (eGFP gene) expression, and they tested whether the T7 RNA polymerase promoter system can regulate gene expression. As the result, fluorescent eGFP gene was successfully reported the promoter’s function in mammalian cells, suggesting that the T7 promoter reporter system helps initiate expression of the eGFP reporter gene. All the above studies suggest that T7 promoter reporter system can be helpful in controlling gene expression.
This information will lead us to further study of the promoter reporter system mechanism for molecular study of disease control and drug development. For example, by controlling the expression of gene, we can control the potential development of breast cancer. The mammalian CYP19 gene (Bulun et. al., 2003), which controls the breast adipose tissue development of women. Under normal condition, CYP19 gene expression is maintained at a low level via promoter I.4. In contrast, increased expression via Promoters II and I.3 activities will cause breast cancer. Reporter promoter system plays an important role in determining promoter function and restore reporter gene expression.
In conclusion, the promoter reporter system is significant in monitoring gene expression. The technique from this project of cloning a promoter to a promoterless plasmid can be used in identifying promoter’s function, and further develop to a molecular method of disease control.
ACKNOWLEDGEMENT:
We would like to thank Dr. Liane Chen for the compilation of all the experimental methods used in this cloning project as well as the design of the LCT7 primer set. We would like to thank Dr. Maryam Moussavi for providing lecture and lab instructions. We would also like to thank Masha Eslami and Ritika Bhinder for the creation of ME/RB primer set. We thank Ryan Ghorayeb and Elizabeth Samuels for providing the transformation control plates and Jade Shivak and Emma Laqua for providing their plasmid screening result. We also thank Nicholas Strowbridge and Yihan Wu for providing help and materials necessary during labs for this project and their feedback on each assignment.
| a) | pGFP-S12 | pGEM-T | ||||
| Restriction Endonuclease | BamHI | NdeI | PvuII | BamHI | NdeI | PvuII |
| Position of Restriction site(s) (bp) | 70 & 614 | 324 & 1088 | No cut | No cut | 82 | 326 & 2890 |
| Predicted Fragment Size (bp) | 544 & 2569 | 764 & 2376 | 3140 | 3000 | 3000 | 436 & 2564 |
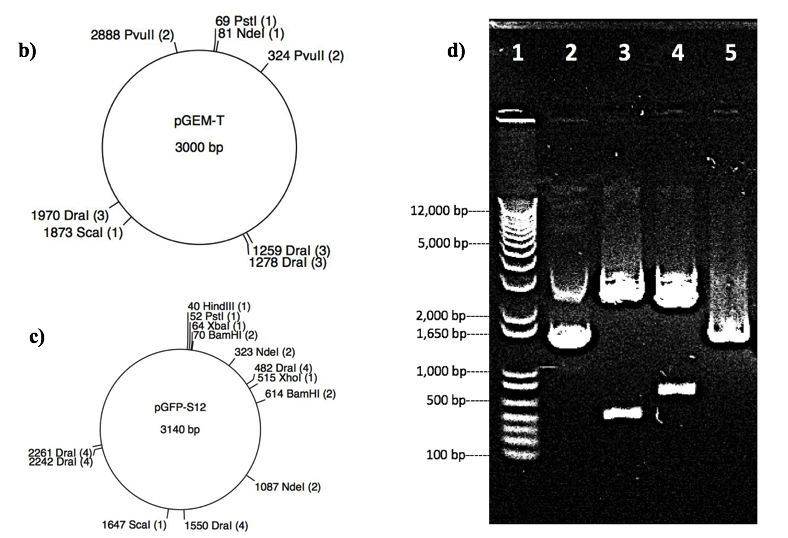
Figure 1: Restriction analysis to determine the identity of unknown plasmid X. a). Table of selected restriction digests (with enzymes BamHI, NdeI and PvuII) and their predicted site(s) and fragment(s) for both PGFP-S12 and pGEM-T. Numbers represent the location or fragment size in basepairs, “N/A” means plasmid remains uncut. b) and c). Map representations of the plasmids pGEM-T and pGFP-S12, respectively, displaying the location of restriction endonucleases for the following enzymes: BamHI, DraI, HindIII, NdeI, PstI, PvuII, ScaI, XbaI, XhoI. Restriction sites are only represented if they are present in the plasmid sequence. d). Gel electrophoresis of plasmid X in 0.8% agarose gel, image shows the relative size of fragments after digestion by BamHI, NdeI and PvuII. Lane number labelled at the top, fragments size indicated on the left. Lane 1 represents 1 Kb invitrogen DNA ladder used to measure fragment size and lane 2 represent uncut plasmid X (control group). Lanes 3, 4 and 5 show plasmid X with restriction enzymes BamHI, NdeI and PvuII, respectively.
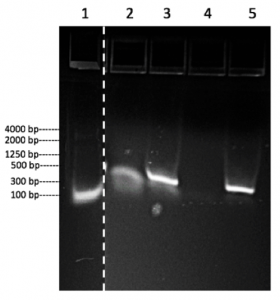
Figure 2: PCR amplification result of the T7 promoter from the pGEM-T plasmid presented on a Lonza FlashGel. Lane number labeled at top, band size labeled at left. Lane 1 shows the Lonza FlashGel standard DNA marker with base pair range from 100 bp – 4000 bp. Lane 2 shows the no template control with the LCT7 primer set. Lane 3 shows the PCR products with the LCT7 primer set. Lane 4 shows the no template control with ME/RB primer set. Lane 5 shows the PCR products with ME/RB primer set.
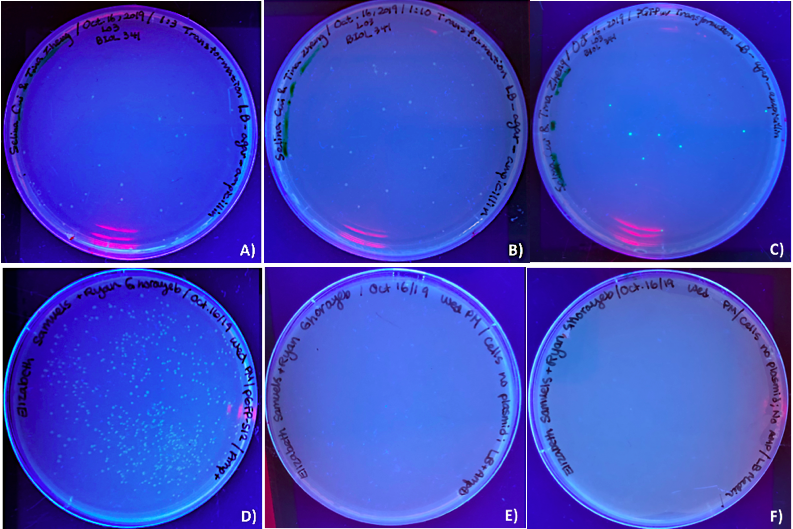
Figure 3. Ultraviolet luminescence of DH5α E. coli cells transformed with ligation of pGFP-S12 with T7 promoter form PCR product. DH5α E. coli cells cultures plated on LB-agar-ampicillin plates (A to E) and plated on LB-agar plates (F) after transformation with the ligation products (T7 promoter region from PCR product) and pGFP-S12 plasmid in two treatments: (A) 1:3 vector to insert ratio and a (B) 1:10 vector to insert ratio. Four control samples were used: (C) E. coli cells transformed with pGFPuv on LBagar-ampicillin, (D) E. coli cells transformed with pGFP-S12 on LB-agar-ampicillin, (E) E. coli cells untransformed on LB-agar-ampicillin and (F) E. coli cells untransfected on LB-agar without ampicillin. Figures 3 D), E) and F) were volunteer samples from Ryan Ghorayeb and Elizabeth Samuels.
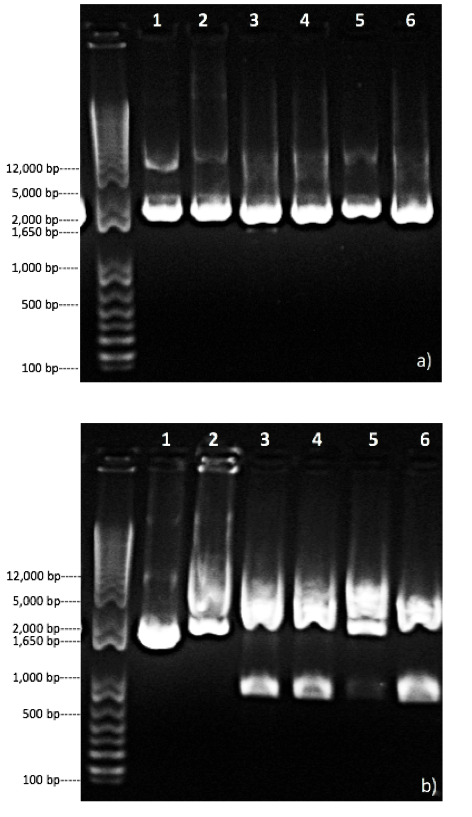
Figure 4. Gel electrophoresis result of structure identification of recombinant plasmid. The leftmost lanes in both figures represent the 1 Kb invitrogen DNA ladder. a) Restriction digest analysis of pGFP-S12 and recombinant plasmid on 1.0% agarose gel after digestion by BglII. Lane 1 represents uncut pGFP-S12. Lanes 2 shows pGFP-S12 digested with enzymes BglII. Lanes 3 and 4 represent the recombinant plasmid from two different 1:3 ligation samples digested with enzymes BglII. Lanes 5 and 6 represent the recombinant plasmid from two different 1:10 ligation samples digested with enzymes BglII. b) Restriction digest analysis of pGFP-S12 and recombinant plasmid on 1.0% agarose gel after digestion by NdeI. This result was provided by Jade Shivak and Emma Laqua. Lane 1 represents uncut pGFP-S12. Lanes 2 shows pGFP-S12 digested with enzymes NdeI. Lane 3 to 6 represent the recombinant plasmid from 4 different ligation samples digested with enzymes NdeI.
REFERENCE
Brogan, J., Li, F., Li, W., He, Z., Huang, Q., & Li, C. (2012). Imaging molecular pathways: Reporter genes. Radiation Research, 177(4), 508-513.
Bulun, S. E., Sebastian, S., Takayama, K., Suzuki, T., Sasano, H., & Shozu, M. (2003). The human CYP19 (aromatase P450) gene: update on physiologic roles and genomic organization of promoters. The Journal of steroid biochemistry and molecular biology, 86(3-5), 219-224.
Chen, L. (2019). Lab Protocols [Lecture Notes]. Retrieved from https://connect.ubc.ca
doi:10.1667/RR2918.1
Ghaderi, M., Sabahi, F., Sadeghi-Zadeh, M., Khanlari, Z., Jamaati, A., Mousavi-Nasab, D., ... & Fazeli, M. (2014). Construction of an eGFP expression plasmid under control of T7 promoter and IRES sequence for assay of T7 RNA polymerase activity in mammalian cell lines. Iranian journal of cancer prevention, 7(3), 137.
Fazeli, M. (2014). Construction of an eGFP expression plasmid under control of T7 promoter and IRES sequence for assay of T7 RNA polymerase activity in mammalian cell lines. Iranian journal of cancer prevention, 7(3), 137.
Goh, E.B., Yim, G., Tsui, W., McClure, J., Surette, M. G., & Davies, J. (2002). Transcriptional modulation of bacterial gene expression by subinhibitory concentrations of antibiotics. Proceedings of the National Academy of Sciences, 99(26), 17025–17030. doi: 10.1073/pnas.252607699
Heifetz, P. B. (2000). Genetic engineering of the chloroplast. Biochimie, 82(6-7), 655-666.
Hoffman, R. M. (2015). Application of GFP imaging in cancer. Laboratory Investigation; a Journal of Technical Methods and Pathology, 95(4), 432. doi:10.1038/labinvest.2014.154
Karunakaran, R., Mauchline, T. H., Hosie, A. H., & Poole, P. S. (2005). A family of promoter probe vectors incorporating autofluorescent and chromogenic reporter proteins for studying gene expression in Gram-negative bacteria. Microbiology, 151(10), 3249-3256.
Kunert, A., Hagemann, M., & Erdmann, N. (2000). Construction of promoter probe vectors for Synechocystis sp. PCC 6803 using the light-emitting reporter systems Gfp and LuxAB. Journal of microbiological methods, 41(3), 185-194.
Naylor, L. H. (1999). Reporter gene technology: the future looks bright. Biochemical pharmacology, 58(5), 749-757.
pathways: Reporter genes. Radiation Research, 177(4), 508-513.
Pike, M. C., Spicer, D. V., Dahmoush, L., & Press, M. F. (1993). Estrogens, progestogens, normal breast cell proliferation, and breast cancer risk. Epidemiologic reviews, 15(1), 17.
QIAGEN. (2019). QIAprep® Miniprep Handbook. QIAGEN.
QIAGEN. (2019). QIAquick® Spin Handbook. QIAGEN.
Reed, M. L., Wilson, S. K., Sutton, C. A., & Hanson, M. R. (2001). High‐level expression of a synthetic red‐shifted GFP coding region incorporated into transgenic chloroplasts. The Plant Journal, 27(3), 257-265.
Scherczinger, C. A., Ladd, C., Bourke, M. T., Adamowicz, M. S., Johannes, P. M., Scherczinger, R., & Lee, H. C. (1999). A systematic analysis of PCR contamination. Journal of forensic sciences, 44, 1042–1045.
Soboleski, M.R., Oaks, J., & Halford, W.P. (2005). Green fluorescent protein is a quantitative reporter of gene expression in individual eukaryotic cells. FASEB Journal, 19(3), 440-442. doi: 10.1096/fj.04-3180fje
Tabor, S. (1990). Expression using the T7 RNA polymerase/promoter system. Current protocols in molecular biology, 11(1), 16-2.
Tabor, S., & Richardson, C. C. (1985). A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proceedings of the National Academy of Sciences, 82(4), 1074-1078.
Waxman, D.J., & Strominger, J.L. (1983) Penicillin-binding proteins and the mechanism of action of beta-lactam antibiotics. Annual Review of Biochemistry, 52, 825-869. doi: 10.1146/annurev.bi.52.070183.004141
Zawadzki, V., & Gross, H. J. (1991). Rapid and simple purification of T7 RNA polymerase. Nucleic Acids Research, 19(8), 1948–1948. doi:10.1093/nar/19.8.1948
Learning Significance